.jpg?height=100&name=IMG_0068%20(1).jpg)
We get a lot of great feedback on our classification tools. In particular, our germline classifier is a tool that healthcare professionals around the world constantly rely on to help them solve cases. This is because it implements recognized industry standard variant interpretation guidelines developed by the American College of Medical Genetics and Genomics. That means VarSome does the heavy lifting of applying the rules in real-time to all of the relevant evidence found in the impressive VarSome aggregated database, giving the user more time to spend understanding the classification in the context of their case.
Richard Meyer was the Chief Technology Officer responsible for building the VarSome classifiers into what they are today, including the implementation of the points-based system seen in the current germline classifier.
Why was it important to have interpretation guidelines, and what does that say about the field today?
RM: The original guidelines published in 2015 by Sue Richards et al. were critical in setting a standard that has been adopted worldwide. There have since been refinements in different countries, but the core methodology, the rule names and the five pathogenicity classes are universally accepted.
These guidelines have then formed a basis for expert panels, including from ClinGen, to then refine or clarify the original proposal, and create additional guidelines for specific diseases, all of this based on feedback from the genomics field, researchers and variant curators.
The recent Tavtigian paper on in-silico predictions and the points-based score have together had a huge qualitative impact on variant classification, providing a solid statistical framework for evaluating the accuracy of individual rules.
Which guidelines have we implemented?
RM: There are two main challenges with automating the guidelines: access to reliable sources of data, and translating the rules into efficient, accurate algorithms.
VarSome integrates a vast number of curated genomics databases, and this has allowed us to implement all the rules. Some data is still difficult to obtain, for example disease and population specific incidence and penetrance statistics.
A huge amount of work has gone into translating concepts such as rule PM1 “located in a mutational hot spot and/or critical and well-established functional domain” into a statistically validated algorithm; we have frequently used machine learning techniques in the process.
As it stands, only the rules related to complex cohorts are not automated in VarSome’s implementation, and cis/trans rules.
How do you approach automating the application of those guidelines?
RM: The first step is to understand what the particular guideline is trying to achieve, what it means biologically, and any caveats or exceptions that need to be considered. The next step is to determine which sources of data will be required to evaluate the corresponding rule, and then obtaining or licensing that data, incorporating it into VarSome’s high performance “MolecularDB”.
A critical final step is validating the implementation: this is performed by comparing the automated classifier to as many curated data-sets as possible, most notably the growing set of 2.1M variants reported in ClinVar. In some cases, numeric methods are employed to calibrate the implementation, extract confidence levels and thresholds - this is where the Tavtigian paper standardizing in-silico calibration comes in very helpful.
What are the major benefits of automating this?
RM: Beyond the time-saving and high-quality aspects of VarSome’s automated classifier, we know that our users value the VarSome community contributions, the aspect that this tool is used by thousands of professionals daily. These users can manually classify variants, link publications, and most importantly feedback any improvements which we aim to implement as soon as possible.
VarSome’s automated classifier is continuously improved and maintained by a team of experts: we calibrate and validate the thresholds for all the in-silico predictors, we review all the data sources used and update them regularly, and in some cases, for example rule PM1 mentioned above, we rigorously codify and validate a somewhat vague concept.
All of these aspects contribute to building a uniquely established and reliable worldwide resource.
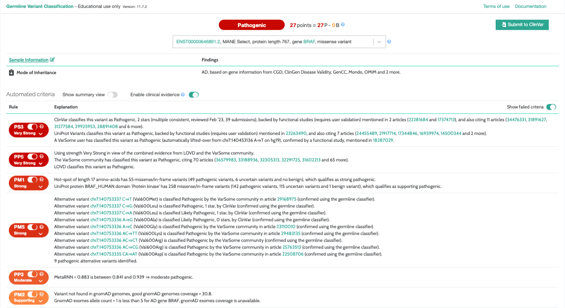
Why was it important for the VarSome classifiers to not be a 'black box' but to clearly show which rules were triggered and the supporting evidence?
RM: From the very beginning it was clear to us that users would not want 'another black box'. We needed total transparency in order for our automated classifier to be accepted by the genomics community. For that reason we have always provided full plain English explanations for every rule triggered, along with evidence used and where it was sourced from.
The VarSome 'Cards User Interface' also makes it very easy and transparent to verify that data themselves, and even, wherever possible, link back to the source web-site. Our documentation also lists in great detail how the rules are implemented. This transparency not only contributes to the platform’s global recognition, but it’s also critical for us to maintain and validate the platform: for every rule you can not only see why it was triggered, but you also get explanations for why it was not triggered - these can be vital in understanding edge cases or problem variants.
If you'd like to learn more about how you can use our germline classifier in VarSome Clinical to support the identification of your variants of interest, reach out to us at sales@VarSome.com.
 Richard Meyer, former Chief Technology Officer in Saphetor SA.
Richard Meyer, former Chief Technology Officer in Saphetor SA.
Richard Meyer is the former CTO in Saphetor SA. Richard has had a distinguished career in the financial technology serving in senior positions in Salomon Brothers/Citigroup, JP Morgan and Deutsche Bank, where he has successfully run global teams of up to 300 engineers in investment banking, electronic trading and risk management across all asset classes. More recently and prior to joining Saphetor, he began a second career in innovative companies. He worked in the music technology business, developing professional DSP synthesisers, including a mobile app showcased by Apple. Richard holds a master’s degree in computer science from the University of Paris. He offers a rare combination of advanced managerial skills, hands on software engineering and an insatiable appetite to learn and solve interesting problems.
About VarSome Clinical
VarSome Clinical is a variant annotation and interpretation platform. Starting from FASTQs or VCFs, VarSome Clinical gives you the ability to analyze whole genomes, exomes, and gene panels in a matter of minutes, with liability and always in control of your results. Get in touch today to find out more, at sales@VarSome.com